

Early Days of the P22 Tail Spike Protein and in vivo Protein Folding
David P. Goldenberg
goldenberg@biology.utah.edu
The first year at MIT
I joined the graduate program of the Biology Department at MIT in the fall of 1976. I had just graduated from Whitman College (a small liberal arts college in Walla Walla, WA) with majors in chemistry and mathematics. But, my real interest was biochemistry and what is now called structural biology. At that time, biology as it was taught at Whitman was of the very classic variety and seemed to require a lot of memorization and drawing. Since I wasn’t very good at drawing and was pretty sure that I couldn’t learn all of the stuff that was required of a biology major, I had chosen to focus on the chemical side of biochemistry. The math major came about because I enjoyed the subject and was especially fascinated by the ways that it could be applied, especially in physical chemistry, which seemed to be the part of chemistry and biochemistry that I was most drawn to. In my senior year, I learned about X-ray crystallography, and that seemed like an ideal way to combine my various interests. One of my professors at Whitman told me that I should apply to MIT for graduate school and work with Alex Rich, whose lab had determined the structure of a tRNA molecule, a landmark accomplishment. To my surprise, I was accepted at MIT, and the opportunity to move from the quiet, sheltered life of Whitman and Walla Walla to MIT, Cambridge and the Boston area was irresistible.
My first year at MIT was eye-opening in almost every respect. The first year of grad- uate studies in the Biology Department was centered around a set of rigorous courses in biochemistry, genetics and molecular biology. (Something that I am very pleased to see has not changed.) Although I felt confident in my undergraduate education in the fundamentals of chemistry and math, the only biology course I had taken (other than biochemistry, which was definitely a chemistry course) was introductory genetics. Suddenly, I was surrounded by a cohort of extremely smart students from prestigious institutions, and they all knew a lot more than I did. I was intimidated, but also excited by the new ways that I was learning to think about scientific problems and how to address them. Molecular biology was presented not so much as a description of what molecules do in a cell, but as an approach to learning what they do and how they do it. In particular, molecular biology at MIT was viewed as a merging of biochemistry and genetics, which provided complementary methodologies and intellectual frameworks that could be used to unravel a variety of molecular mechanisms. This was heady stuff, and my classmates and I were caught up in the excitement of rapid new advances, especially the development of recombinant DNA technology and the controversies that it engendered (in which Jon played a very visible role).
Also during that first year, I started to have doubts about my original plan to pursue X-ray crystallography. As I talked to the members of Alex Rich’s lab, I learned how slow progress in this field was and was told, in essence, that there was no future in it. When I now teach undergraduates about crystallography, I show the graph in Fig 1, which plots the growth of the Protein Data Bank (PDB) from 1976 (the year in which the PDB became publicly available and I started graduate school) to 2020. Obviously, there was a future in crystallography!
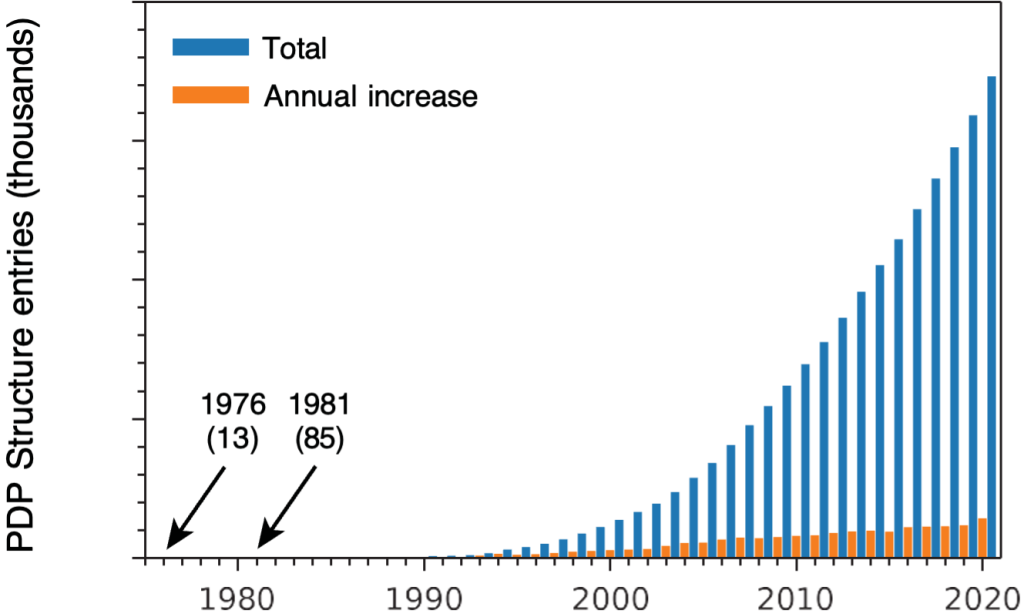
But, I also point out to students that between 1976 and 1981, when I finished my Ph.D., there were only 72 structures added to the PDB, and the odds that I would have determined one of them were not all that good. In any event, I started to think about other kinds of research projects.
Unlike many biology graduate programs today, the MIT program didn’t include laboratory rotations for first-year students. Instead, all of the faculty members made presentations about their research during the independent activities period, a few weeks between the fall and spring semesters. There was a lot to absorb over a short time, and I am sure that most of it went past me. But, Jon King’s presentation drew my full attention. By now, I could ap- preciate the ability of genetic analysis to dissect molecular processes, and Jon’s elegant work on the assembly of bacteriophages, which also drew on biophysical separations and electron microscopy, was enormously appealing. He also described a new project in which he planned to use temperature-sensitive (ts) mutations to study the processes by which proteins fold into their three-dimensional structures. During the fall semester, I had taken a wonderful course in physical biochemistry (taught by Phil Sharp and Paul Schimmel) and had learned about experimental and theoretical approaches to understanding protein folding, so I was primed to hear about this. The general idea of using mutations to study protein structure and function was in the air, but hadn’t been developed very far yet, and Jon’s focus on the folding process was especially exciting to me. There is also no denying that his charisma and lucid presentation helped draw me in. During the following semester, I took Jon’s course on morphogenesis (molecular assembly, really), and the hook was firmly set. At the end of the semester, we agreed that I would join Jon’s lab for my Ph.D. research.
But, I hadn’t given up my interest in protein structures as determined by X-ray crystallography. In my reading, I had learned that the three-dimensional structure of phage T4 lysozyme had been determined a few years earlier in Brian Matthews’ lab (1). Brian was (and remains) a member of the Institute of Molecular Biology at the University of Oregon, which also included George Streisinger, who had carried out a detailed genetic analysis of lysozyme. It was clear from this first paper that the intention was to combine genetics and crystallography to probe the determinants of protein structure and function, a program that blossomed several years later, with my MIT classmate, Tom Alber, playing a major role (2). At the time, it seemed to me as though Jon and I should jump in and use T4 lysozyme to study folding, and he seemed to go along with this idea when I joined the lab. But, he quickly convinced me that it would be much more interesting to study a phage structural protein, which was his intention all along!
Temperature-sensitive mutants and protein folding
During my first year in the lab, I worked on a phage T4 structural protein, the product of gene 12 (gp12), which forms short fibers on the tail baseplate. Several years before, Jon and Ulrich Laemmli had shown that the maturation of this protein was dependent on the activity of another protein, gp57, which also plays a role in the assembly of the long fibers (3). This observation, and the presence of multiple ts mutations in gene 12 led Jon to believe that there would be interesting things to be learned about the folding of this protein. Under the tutelage of Peter Berget, a post-doc in the lab who had purified two other proteins from the T4 baseplate (4), I learned the basics of working with bacteria and phage, starting with a single-burst growth experiment, and then moved on to try to purify gp12 using an in-vitro complementation assay. This did not go well. Although a protocol for purifying the protein had been published by Robert Hasselkorn’s group (5), I found that the protein tended to favor the insoluble fraction of cell lysates, and my attempts to coerce it into a soluble form suitable for chromatography were futile. Many years later, the protein was extensively studied by others; it’s three-dimensional structure was determined; and in vitro folding experiments confirmed that gp37 acts as a chaperone in gp12 folding (6). Jon’s early observations of the effects of gp57 on the maturation of the long and short tail fibers is likely the first intimation of folding chaperones.
Perhaps we gave up on gp12 two soon, but Jon and I agreed that I should shift my attention to the tail spike protein (TSP) of phage P22. The gene encoding the protein, gene 9, was known to be rich in ts mutations, and these were being mapped by Peter and Donna Smith, a talented research technician (7). Phage with these mutations could grow at 30 ◦C, but not at 39 ◦C, and phage particles missing the tail spike accumulated at the restrictive temperature. All of the indications were that the proteins failed to fold at the higher temperature, but there was very little direct evidence. At this time, DNA sequencing was far from routine, and we did not know the sequence of the gene or protein, much less its three-dimensional structure. Mutants were only identified by codes assigned when they were isolated.
A year or two before I joined the lab, Peter and Tony Poteete had worked out an efficient protocol for purifying the tail spike (8), and Peter guided me through the process. It quickly became evident that gp9 of P22 was much easier to work with than gp12 of T4. In addition to the wild-type protein, I purified the proteins encoded by three of the ts mutations, from bacteria infected at the permissive temperature. Peter and Tony had found that the purified wild-type protein was remarkably heat resistant, requiring incubation at 90 ◦C in order to observe inactivation at a significant rate. In order to test whether the ts mutations rendered the proteins thermally unstable, I subjected the mutant proteins to this treatment and found that they were equally heat-resistant. Thus, the failure of the mutant phage to grow at 39 ◦C could hardly be attributed to thermal sensitivity of the folded protein. But, three mutants made for a small sample size, and Jon insisted that I carry out a more extensive set of experiments, with a total of 13 ts mutants, but using crude lysates, rather than purifying all of the proteins. I found that all of the mature proteins displayed the same high thermal stability, and that they were fully functional in binding to phage heads and facilitating adsorption to the bacterial host, at both 30 and 39 ◦C (9).
At about the same time, Donna carried out a set of experiments to characterize the mu- tant proteins synthesized at the restrictive temperature (10). Using a pulse-chase labeling protocol with 14C-labeled amino acids, she established that the mutant polypeptides were synthesized at the same rate as the wild-type at 39 ◦C and accumulated stably. But, the mutant proteins synthesized at the higher temperature were unable to bind to phage heads or to the bacterial O-antigen. Most remarkably, she found that some of the mutant proteins synthesized at 39 ◦C could be rescued if the temperature was subsequently lowered. This result suggested that these proteins represented a state that was on the folding and assembly pathway, or at least not too far off it. I am still embarrassed to recall my resistance to accept- ing this result: Somehow, I had convinced myself that the ability of the proteins synthesized at 30 ◦C to retain their structure and function at 39 ◦C (and much higher temperature) meant that the proteins synthesized at the higher temperature should retain their inactive state if the temperature was lowered. This was theoretical reasoning gone thoroughly amuck, and I learned a lasting lesson about being very cautious about theories, especially my own and especially in the face of experimental data!
Together, Donna’s results and my own showed that the ts mutations in gene 9 prevented correct folding or assembly of the tail spike at 39 ◦C. This was not really an unexpected result, as it had been long known that some ts mutations only affected function if the protein was synthesized at the restrictive temperature. In a classic paper, Sadler and Novick had introduced the term temperature-sensitive synthesis (tss) to distinguish mutations of this class from those that rendered the protein thermolabile (11). Indeed, the knowledge of such mutations was a major factor in Jon’s strategy to use ts mutations to study protein folding. But, Donna’s results allowed us to make the further distinction that the mutations affected folding, rather than biosynthesis of the polypeptide chain, and Jon later introduced the term temperature-sensitive folding (tsf ) to emphasize this distinction.
With this background, the next challenge was to find out what happened to the polypep- tide chains when they folded correctly, or failed to. This led to an attempt to set up an in vitro translation system that could be used to monitor folding during or shortly after syn- thesis. I had raised antibodies in rabbits that were specific for native and denatured forms of the protein (my first and only experience with animal research), and the plan, as best I recall it, was to use these antibodies to characterize the translation products. Unfortunately, my attempts at in vitro protein synthesis didn’t get very far and were quickly abandoned.
At some point, Jon and I started thinking about using in vivo pulse-chase labeling to study the kinetics of folding. Margaret (Minx) Fuller, a more senior graduate student in the lab (who went on to a distinguished career in developmental biology), had been trying to study P22 capsid assembly this way, These experiments used the strategy that Jon and Yoshiko Kikuchi had so successfully employed to study assembly of the T4 baseplate (12), which involved fractionating pulse-labeled cell extracts by sucrose-gradient centrifugation and then analyzing the gradient fractions by SDS-gel electrophoresis. Although Minx wasn’t able to detect in vivo capsid-assembly intermediates, she made an observation that proved critical to my work: Immediately after biosynthesis, gp9 polypeptide chains accumulated in a rapidly sedimenting form, associated with cellular debris, but then appeared in a soluble form with the sedimentation rate of the native protein. Because the pulse-chase time intervals were relatively long (on the order of minutes), these results suggested that whatever processes were required for the maturation of TSP, they were not so fast as to preclude analysis by relatively simple biochemical or biophysical techniques. A stopped-flow spectrometer was not required!
Rather than trying to follow up Minx’s experiments with sucrose gradients, I began to fool around with different forms of electrophoresis, inspired in large part by the work of Tom Creighton, who was using electrophoresis to separate disulfide-bonded intermediates in the folding of bovine pancreatic trypsin inhibitor (BPTI). In their earlier work with TSP, Peter and Tony discovered that the mobility of the protein in SDS gels depended on whether or not the sample was heated in a boiling water bath, with the un-heated protein migrating much more slowly than what was presumed to be the fully denatured and dissociated form. This appeared to be an easy way to distinguish the fully folded proteins from other forms, and I used this assay as the basis for a pulse-chase labeling experiment. The experiment was very simple: Bacteria were infected with phage deficient in head-protein assembly (so that mature tail spikes would not assemble onto capsids) and pulse-labeled for 2 min with 14C- labeled amino acids, followed by unlabeled amino acids. Samples were quickly withdrawn; lysed by freezing and thawing; mixed with SDS-containing buffer (but not boiled) and elec- trophoresed, followed by autoradiography of the gel. Rather remarkably, this experiment worked the first time I tried it, and the gel shown in the resulting paper is the one from that first experiment (Fig. 2).
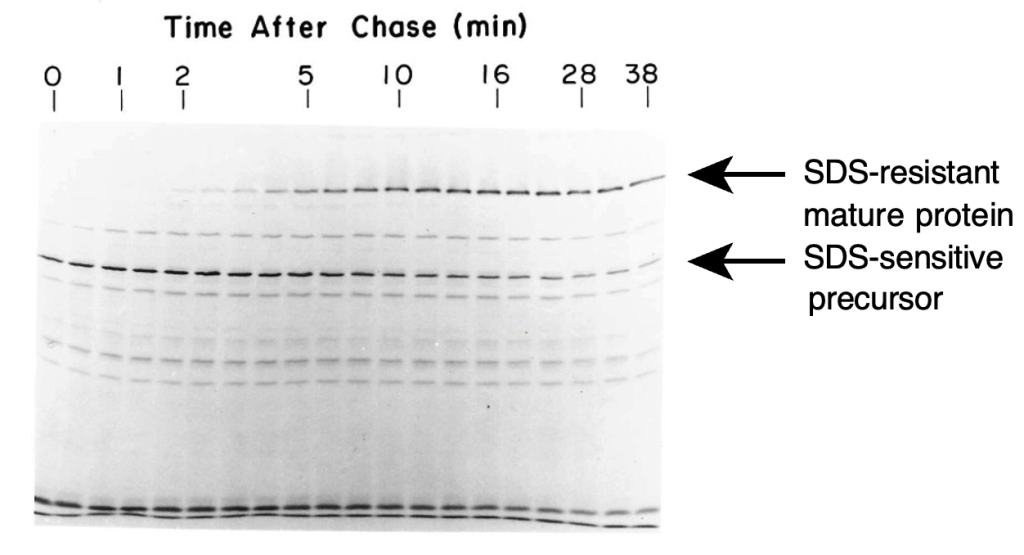
I still vividly recall developing the autoradiogram in the tiny (unventilated) lab darkroom and seeing the bands appear even before the film was cleared in the fixer solution. The autoradiograph clearly showed the conversion of an SDS-sensitive form of the polypeptide chain to the mature SDS-resistant form, with a half-time of about 5 min. Although this was not the first experiment to examine protein folding in vivo, it was certainly among the first, and I remember hearing some grumbling from the more traditional protein chemists, along the lines of “WHAT is protein folding in vivo?”, when I presented the results at a Proteins Gordon Conference in 1981. Now, of course, it is commonplace to examine the intracellular fate of newly synthesized proteins.
This initial experiment led to a rapid succession of others, over a short period of time that I remember as one of the most exciting and productive in my career. In addition to the trick of using unboiled samples for SDS gels, I experimented with electrophoresis without SDS at all, that is non-denaturing (or native) gel electrophoresis, where proteins migrate on the basis of their own intrinsic charge.1 Using these gels, I discovered that some of the ts mutant proteins (synthesized at the permissive temperature) displayed slightly altered mobilities, suggesting that they contained amino-acid replacements that altered the electric charge of the protein. When bacteria were co-infected with phage carrying the wild-type gene and phage with one of these mutations, four distinct species could be detected on the gels. This was strong evidence that the protein was a trimer, with four bands representing the two kinds of homotrimers and two kinds of heterotrimers. At that time, measurements of the molecular weights of the polypeptide chain and the mature tailspike had given ambiguous results regarding the quaternary structure. The results also argued against models in which the trimers formed by association between chains still being synthesized on the same polysome.
In the hopes of resolving intermediates in production of the native trimer, I then tried using non-denaturing gels to analyze samples from pulse-labeled extracts. For these exper- iments, the samples were kept on ice and the gels were run in the lab’s cold room. In the resulting gels, the formation of the native protein could again be seen, with the same kinet- ics as observed with the SDS-gels of unheated samples, and we could also see a more slowly migrating species that appeared and then disappeared as the native trimer appeared. The kinetic properties of this species (and the fact that it did not appear in lysates of bacteria infected with phage carrying amber mutations in gene 9), strongly suggested that it was an intermediate in the formation of the native trimer. There were also broader bands that appeared transiently and migrated more rapidly than the native trimer, but the characteri- zation of these was left to others.
Because the newly identified species migrated more slowly than the native trimer, Jon and I suspected that it might be a trimer (or perhaps dimer) in which the chains had associated but hadn’t completely folded. Unfortunately, the band formed by this species wasn’t very sharp, and the resolution of the non-denaturing gels wasn’t sufficient to determine the quaternary structure of this species directly using the charge-change variants. But, I was able to show that it was most likely a trimer in a slightly round-about way. After lysing the bacteria at low temperature, a fraction of the molecules could be converted to the native form after being warmed to 30 ◦C. When the lysate used for this reaction came from bacteria co-infected with phage encoding the wild-type protein and a charge-change variant, the characteristic pattern of four bands was seen for the native protein produced in the in vitro reaction. If, however, separate lysates containing the two kinds of polypeptides were mixed before being warmed, only homotrimers were produced, indicating that the chains had already associated before the samples were mixed. I was also able to fractionate the lysate on a gel-filtration column and show that the molecules that could be converted to the native trimer had dimensions that were similar to those of the native protein. Based on this evidence, Jon and I felt confident that the precursor was trimeric and dubbed it the protrimer, in obvious homage to the pro-capsid intermediates in the assembly of double- stranded DNA viruses (13).
For the final set of experiments for my Ph.D. thesis, I returned to the ts mutants and asked whether or not they could form the protrimer at the restrictive temperature, using the same pulse-chase labeling protocol and non-denaturing gel electrophoresis described above. Although we probably hoped that some of the mutations would be shown to act before formation of the protrimer and some after, we found that all of them prevented formation of this intermediate (15). The picture that emerged from these results was one in which the structure of a precursor to the protrimer, likely a partially folded monomer, was particularly sensitive to amino-acid replacements and elevated temperature. In the paper reporting these results, we also hypothesized that the subunits of the native trimer might be somehow intertwined, thereby enhancing the stability of the native protein, but requiring the existence of a partially folded trimeric intermediate. That prediction was based on the scantiest of evidence, but was found to be correct when the crystal structure of the native protein was determined more than ten years later (16).
For nearly 30 years after I left MIT, Jon’s lab (and others) continued to study the P22 tailspike protein, revealing additional details of the folding and assembly pathway, and the non-productive pathways leading to aggregation, as well as its function as a cell-adhesion molecule. It has been a continuing delight to see new discoveries emerge from the study of this remarkable protein!
Beyond the special case of the tailspike, the general subject of protein folding, as it occurs in vivo and is coupled to other processes, has blossomed into an vibrant area of research, with implications for health, agriculture and biotechnology. When Jon’s lab began its work on ts mutations, there were only a handful of labs in the world working on protein folding, and nearly all of them were studying the in vitro unfolding and refolding of small proteins, such as ribonuclease A and BPTI. I was quite enamored of this literature, but Jon clearly understood (and repeatedly reminded me) that there was much more to be learned about folding than could come from that type of experiment. That view has been spectacularly validated over the past four decades!
The journey after MIT
In the summer of 1979, I attend the XIth International Congress of Biochemistry in Toronto, Canada and presented a poster describing our early work on TSP and the ts mutants, my first presentation at a scientific meeting. This was about a year before the first of the pulse- labeling experiments, and it wasn’t very clear at that point how I would produce a Ph.D. thesis, but for some reason I was already thinking about a postdoctoral position. One of the featured speakers at the conference was Tom Creighton, from the Medical Research Council Laboratory of Molecular Biology (LMB) in Cambridge, England. I had been following Tom’s work on the disulfide-coupled folding of BPTI for some time, and I was anxious to meet one of my scientific heros. He visited my poster, and, with some trepidation, I asked about the possibility of doing a postdoc with him. As with my admission to MIT, I was pleasantly surprised when this became a reality.
Two years later, with the support of an NIH postdoctoral fellowship, I moved to the other Cambridge and began working with Tom on BPTI folding. I spent four years at the LMB, and the experience that I gained there was an ideal complement to the training I received in Jon’s lab. From studying mutant proteins in vivo, I moved to preparing and characterizing chemically modified proteins and determining folding mechanisms in vitro, using the elegant disulfide methods that Tom had developed. The scientific environment of the LMB was always stimulating, and I spent many happy hours in the canteen learning from the other members of the Structural Studies division. In my last year at the LMB, I began work to construct a plasmid to produce BPTI in E. coli, using a gene that had recently been cloned by Barry Kingston and Steve Anderson (17). This was early days for this sort of thing, and progress was very slow, but I was able to learn the basics of DNA manipulation, including site-directed mutagenesis as it was then practiced, which helped lay the foundation for a mutational study of BPTI folding when I started my own laboratory.
In the summer of 1985, I moved back across the Atlantic to take a faculty position in the Biology Department (now known as the School of Biological Sciences) of the University of Utah, in Salt Lake City. I count this as the third big lucky break in my career, after MIT and the LMB, as it again placed me in a great environment with wonderful colleagues. The department had been formed in the early 1970s by the merger of multiple more specialized departments, under the leadership of K. Gordon Lark. Like Jon, Gordon was a product of the phage group that surrounded Max Delbru¨ck and other pioneers of molecular biology during the 1950s and 60s, and he made a point of bringing the ethos of that group, with its emphasis on collegiality and mutual respect, to the department. (With varying degrees of success, of course!)
After a few months spent ordering supplies and equipment, I renewed my efforts to produce BPTI in bacteria, and before too long was able to begin studying the folding of variants with engineered amino-acid replacements. This evolved into a long-term project that was very much a melding of the approaches that I learned from Jon King and Tom Creighton, combining mutational analysis with biochemical and biophysical approaches. I think that this project also reflected a general approach to science exemplified by my two mentors: Dig into the details of a complicated process and a big picture will emerge. In this case, our systematic analysis of the effects of amino-acid replacements on each step in the BPTI folding pathway led to a general picture of how individual interactions contribute cooperatively to stability as structure forms during folding (18).
The studies of disulfide-coupled folding of BPTI were later extended to work on cono- toxins, small disulfide-bonded peptides discovered by my Utah colleague Baldomero (Toto) Olivera (19). The BPTI variants also led us to examine the question of why the protein is, in fact, an inhibitor of trypsin and other proteases, rather than a substrate. This question led my group to use NMR spectroscopy, through a collaboration with Terry Oas at Duke University, to study the effects of amino-acid replacements on protein dynamics and flexi- bility (20). Rather than being content to be a user of NMR methods, my old inclinations towards physics and math drew me to a deep dive into the mysteries of just how NMR works and the underlying quantum mechanics. After spending an unreasonable amount of time on the principles of NMR, I felt somewhat obliged to write down what I had learned, in the hopes that it would help others, resulting in a textbook on the subject (21). My lab even managed to do a little bit of X-ray crystallography, in collaboration with Martin Horvath, finally enabling me to contribute a few crystal structures to the Protein Data Bank (22)! The last major area that my group investigated was the behavior of unstructured proteins, especially in the presence of high concentrations of globular proteins, using small-angle X-ray and neutron scattering (SAXS and SANS) (23).
Although my lab group was never very large, I was fortunate to attract a number of talented graduate students, research technicians and postdocs, as well as some truly excep- tional undergraduates, to join in these efforts. Of particular note are Scott Beeser, James Berger, Grzegorz Bulaj, Lonnie Coplen, Gretchen Domek, Mike Hanson, Daniel Laheru, Jose Mendoza, Marian Price-Carter, Jian-Xin (Jix) Zhou and Elena Zakharova. I am immensely grateful to these individuals, and many others, for the privilege of working with them and the important contributions that they made to our research. I would like to think that I passed on at least a little of what I learned from Jon!
Notes
- In a 2022 JBC Reflections paper, Jon describes how Uli Laemmli, with help from Jake Maizel and Jon, adapted the discontinuous buffer system of Davis and Ornstein for electrophoresis in the presence of SDS (14). By the time that I was a graduate student, less than a decade later, SDS gels had almost completely replaced the non-denaturing flavor of electrophoresis in most laboratories, and I was, in a sense, taking a step backwards by removing the SDS!
References
- Matthews, W. & Remington, S. J. (1974). The three dimensional structure of the lysozyme from bacteriophage T4. Proc. Natl. Acad. Sci., USA, 71, 4178–4182.
https://doi.org/10.1073/pnas.71.10.4178 - Alber, & Matthews, B. W. (1987). Structure and thermal stability of phage T4 lysozyme. Methods Enzymol., 154, 511–533.
- King, & Laemmli, U. K. (1971). Polypeptides of the tail fibres of bacteriophage T4.J. Mol. Biol., 62, 465–477.
http://dx.doi.org/10.1016/0022-2836(71)90148-3 - Berget, P. B. & King, J. (1978). Isolation and characterization of precursors in T4 baseplate assembly: The complex of gene 10 and gene 11 products. Mol. Biol., 124, 469–486.
http://dx.doi.org/10.1016/0022-2836(78)90182-1
- Mason, S. & Haselkorn, R. (1972). Product of the T4 gene 12. J. Mol. Biol., 66, 445–469.
https://doi.org/10.1016/0022-2836(72)90426-3 - Miller, & Burda, M. R. (1999). Folding of coliphage T4 short tail fiber in vitro. Eur. J. Biochem., 265, 771–778.
https://doi.org/10.1046/j.1432-1327.1999.00782.x
- Smith, D. H., Berget, P. B. & King, J. (1980). Temperature-sensitive mutants blocked in the folding or subunit assembly of the bacteriophage P22 tail spike I. Fine structure mapping. Genetics, 96, 331–352. https://doi.org/10.1093/genetics/96.2.331
- Berget, P. B. & Poteete, A. R. (1980). Structure and functions of the bacteriophage P22 tail protein. Virol., 34, 234–243.
https://doi.org/10.1128/jvi.34.1.234-243.1980
- Goldenberg, P. & King, J. (1981). Temperature-sensitive mutants blocked in the folding or subunit assembly of the bacteriophage P22 tail spike protein. II. Active mutant protein matured at 30 degrees C. J. Mol. Biol., 145, 633–651. http://dx.doi.org/10.1016/0022-2836(81)90307-7
- Smith, H. & King, J. (1981). Temperature-sensitive mutants blocked in folding or subunit assembly of the bacteriophage P22 tail spike protein. III. Inactive polypeptide chains synthesized at 39oC. J. Mol. Biol., 145, 653–676. http://dx.doi.org/10.1016/0022-2836(81)90308-9
- Sadler, R. & Novick, A. (1965). The properties of repressor and the kinetics of its action. J. Mol. Biol., 12, 305–327.
- Kikuchi, Y. & King, J. (1975). Genetic control of bacteriophage T4 baseplate morpho- genesis: I Sequential assembly of the major precursor, in vivo and in J. Mol. Biol., 99, 645–672.
http://dx.doi.org/10.1016/S0022-2836(75)80178-1 - Goldenberg, D. & King, J. (1982). Trimeric intermediate in the in vivo folding and subunit assembly of the tail spike endorhamnosidase of bacteriophage P22. Natl. Acad. Sci., USA, 79, 3403–3407.
http://dx.doi.org/10.1073/pnas.79.11.3403
- King, J. (2022). Using T4 genetics and Laemmli’s development of high-resolution SDS gel electrophoresis to reveal structural protein interactions controlling protein folding and phage self-assembly. Biol. Chem., 298, 102463.
https://doi.org/10.1016/j.jbc.2022.102463 - Goldenberg, D. P., Smith, D. H. & King, J. (1983). Genetic analysis of the folding pathway for the tail spike protein of phage P22. Natl. Acad. Sci., USA, 80, 7060– 7064.
http://dx.doi.org/10.1073/pnas.80.3.760 - Steinbacher, S., Seckler, R., Miller, S., Steipe, B., Huber, R. & Reinemer, P. (1994). Crystal structure of P22 tailspike protein: Interdigitated subunits in a thermostable trimer. Science, 265, 383–386.
http://dx.doi.org/10.1126/science.8023158 - Anderson, & Kingston, I. B. (1983). Isolation of a genomic clone for bovine pancreatic trypsin inhibitor by using a unique-sequence synthetic DNA probe. Proc. Natl. Acad. Sci., USA, 80, 6838–6842.
http://dx.doi.org/10.1073/pnas.80.22.6838
- Bulaj, & Goldenberg, D. P. (2001). ϕ-Values for BPTI folding intermediates and implications for transition states. Nature Struct. Biol., 8, 326–330.
http://dx.doi.org/10.1038/86200 - Price-Carter, , Hull, M. S. & Goldenberg, D. P. (1998). Roles of individual disulfide bonds in the stability and folding of an ω-conotoxin. Biochemistry, 37, 9851–9861.
http://dx.doi.org/10.1021/bi9803978 - Hanson, M., Beeser, S. A., Oas, T. G. & Goldenberg, D. P. (2003). Identification of a residue critical for maintaining the functional conformation of BPTI. J. Mol. Biol., 333, 425–441.
http://dx.doi.org/10.1016/j.jmb.2003.08.023
- Goldenberg, D. P. (2016). Principles of NMR spectroscopy: An illustrated guide. Uni- versity Science Books, Mill Valley, California.
http://www.uscibooks.com/goldenberg.htm - Zakharova, , Horvath, M. P. & Goldenberg, D. P. (2009). Structure of a serine protease poised to resynthesize a peptide bond. Proc. Natl. Acad. Sci., USA, 106, 11034–11039. http://dx.doi.org/10.1073/pnas.0902463106
- Goldenberg, P. & Argyle, B. (2014). Minimal effects of macromolecular crowding on an intrinsically disordered protein: A small-angle neutron scattering study. Biophys. J., 106, 905–914.
http://dx.doi.org/10.1016/j.bpj.2013.12.003