

From Building Model Airplanes to Learning How Viruses and Complex Proteins are Built, at MIT, with Many Partners
Jonathan King
Professor of Molecular Biology, Emeritus
Massachusetts Institute of Technology
Cambridge, MA
jaking@MIT.edu
Early Years in Brooklyn
When I was three years old I contracted polio, affecting my knees. Though I recovered, for some years I was less mobile than my peers, and spent more time indoors than playing in the street. I spent much of that time with Erector sets, Lincoln Logs, and then model airplanes and boats. As a result, I became very interested in how things were built. When the opportunity appeared, I watched construction projects with great interest.
My block in Brooklyn was almost all two-family houses, with the fronts either paved over, or yards that were mostly dirt. But in front of every house was a stately Maple tree.
As a sophomore at James Madison High School I took Biology with Ms. Markhan. She was a warm engaging teacher, and described cogently how the xylem and the phloem in the Maple tree trunks enabled trees to thrive. I was struck at how much was going on behind the bark of those trees. That was the first glimmer of my later interest in Biology.
Both my older brothers, Frank and Steve, had been outstanding basketball players. Trying to follow or limp along that path, I joined the James Madison football team, and played Tight End. From years of reading I had a large vocabulary so scored high on standardized tests. Combining that with playing football landed me a scholarship to Yale.
Entering Biology/Zoology at Yale
At Yale my freshman Science professor was Ernest C. Pollard, who had worked with Rutherford in Great Britain, and subsequently built the first cyclotron in the U.S. He was quite charismatic and a founder of the Biophysics Society, and painted Biophysics as a romantic option, focused for him on interactions of radiation with cells and organisms. Years later his course helped me master electron microscopy, and Biophysics returned to the fore.
One of my Sophomore classes was interdisciplinary Science II, taught by ecologist Edward S.Deevey, Jr. He was a freshwater bog specialist, and the class covered only a very narrow range of material close to his interests. But in the spring, he announced the availability of a summer job assisting him in his research on Connecticut lakes. I was the only student to apply, and thus was accepted.
The project involved driving to local lakes, rowing out toward the middle, and then collecting water samples at different depths. These were returned to the lab, and in the following week analyzed by standard water chemistry techniques (titrating to a color standard) to measure dissolved oxygen and CO2 levels. As the summer progresses, the deep waters get deprived of oxygen, but at some point, in the fall when the surface waters have cooled, the entire water column “turns over” and the deep water gets re-oxygenated.
Deevey was also interested in the composition of the gases bubbling up from the lake bottom mud, and I proudly designed and built a frame which had a gas collecting bottle that we let down to the bottom. The gas bottle could be raised and lowered by a pulley mechanism and the composition of the gas sample determined.
I had very little idea as to what this was all about, but I loved sitting out in the lake in the row boat, while getting paid for the time. Years later I learned that the grant was really to follow radioactive carbon fallout from nuclear weapons testing, which had sunk to the lake bottom, and was retained in the mud and mud gases. This recognition added to my learning to examine very closely the actual goals of research and policy grants, compared to the public relations titles.
At the time I was most interested in psychological and social issues, and initially enrolled in a major then called Culture and Behavior. The material was fascinating but I couldn’t see how one made a living in that arena. One of the courses was a Zoology course, Animal Behavior, taught by lemur specialist Richard Andrews. I knew from the summer job with Professor Deevey that you could make a living as a biological scientist. Yale had separate Zoology and Botany Departments, so I ended up majoring in Zoology.
The course work in Genetics, Biochemistry, Physiology and Embryology, all with two afternoon labs a week was interesting and engaging. Though I didn’t have any professors who knew who I was or cared. The research culture of focusing on graduate students was already in place.
Unfortunately, the labs conflicted with afternoon sports practices. Though Yale had admitted me on the assumption I would go out for Tight End in football, I was pretty sure I didn’t have the talent needed. Instead I joined the Crew and rowed many an afternoon on the Housatonic, which was a good change from classroom pressures. As a sophomore I had a good shot at the Varsity crew. However, I knew I couldn’t make a living following that route. All of my fellow oarsman came from wealthy families, and had no worries about “making a living” after Yale. So, I chose science labs and the academic path.
My genetics professor was a classical geneticist and the lab used Drosophila as the experimental material. But our TA Susan Goldhor (later married to my MIT Nuclear Disarmament physics sidekick Aron Bernstein) gave a few surreptitious lab lectures on the emergence of bacteriophage as an experimental organism for identifying essential genes. I was fascinated. That primed me for the subsequent decision at Caltech to join R. S. Edgar’s T4 laboratory. My classmate Al Kapuler, also from Brooklyn, had published a molecular genetics paper as an undergraduate. When he told me that he was only considering Caltech and Rockefeller Institute, I followed suit. I soon learned of the existence of fellowships to grad school, applied, and was awarded a Woodrow Wilson National Fellowship ($1500-a-year living expenses).
Graduate Studies at Caltech: Genetic Control of T4 Assembly with R.S. Edgar and Bill Wood
When I arrived at Caltech I was shocked to discover it was a tiny campus surrounded by Pasadena residences. There wasn’t even a coffee shop in walking distance, and there was no public transportation. It was almost monastic. On top of that the smog was sufficiently heavy that one couldn’t even see the San Gabriel Mountains that adjoined Pasadena.
I was assigned to work with Professor Alan Hodges, who ran the electron microscope facility, situated in the 2nd basement. Though he had done a brilliant EM analysis with F.O. Schmidt at MIT revealing the fundamental nature of collagen molecules packing into quarter staggered collagen fibrils, I wasn’t going to spend five years in a second basement with no windows.
At Yale, a postdoc had told me that the most interesting work at Caltech was being done by a young faculty member, Robert S. Edgar, who worked on the genetic control of T4 Assembly. In our meeting, Edgar described to me that he was figuring out how organisms got built. That spoke to me and I joined his lab. (Though his lab was in the first basement, there were windows looking up to the street.)
We had a cohort of four students, Dick Russell from Harvard, Bob Huskey from Oklahoma, and Jeffrey Flatgaard from Johns Hopkins; we worked at benches in the center of the room so all worked closely together. Though the four of us were as different as you could imagine, that insured that we formed a lively group, which provided a community for me.
Edgar was painfully shy but he was a brilliant experimentalist who had developed the system of temperature sensitive mutants to map the genes controlling the essential proteins of T4. He was uncomfortable with someone as outgoing as I was, and often avoided me. But I recognized that I could really learn from him, and to not worry about the style of our interactions.
At that time, most of the genes identified in experimental organisms like fruit flies, corn, and Neurospora, controlled morphological features, such as eye color in fruit flies, or shriveled skin in peas, or colony morphology for fungi. Though many essential structural proteins such as actin, myosin, or collagen had been characterized by biochemists, geneticists had not been able to isolate or propagate mutants affecting these proteins.
Edgar’s isolation of conditional lethal temperature sensitive mutants changed this. Mutations lethal at high temperature, could be propagated at low temperature. At the same time Richard Epstein working with Edouard Kellenberger in Geneva, Switzerland had developed the system of amber mutations, which were lethal on one bacterial host, but could be propagated on another host that carried a suppressor mutation. The phage mutations were auxiliary stop codons that prevented translation of the complete protein; the suppressor hosts carried tRNA mutations which generated a modified tRNA that could translate through the stop codon.
These mutations mapped to more than 40 different genes of T4. Genetics with phage was very easy; you mixed two mutant strains together with susceptible bacteria, incubated for an hour, and then plated on host bacteria on Petri dishes, to look for recombinants, which appeared by the following day.
Kellenberger, who was an electron microscopist, showed that the effect of many of these mutations under the restrictive conditions, was to prevent formation or assembly of complete phage particles. They often observed that substructures such as heads or tails accumulated in the mutant infected cells under restrictive conditions. I was struck by the results, which showed clearly that we had access to genes whose proteins were needed to build phage particles.
Tackling Tail Fiber Assembly with Specific Antibodies
Edgar and his talented technician Ilga Lielausis decided to pursue the formation of the long thin phage tail fibers, which were known to be the target of anti-phage antibodies. In a brilliant experimental analysis, they showed that antibody against tail fibers were composed of five classes of antibodies, directed against at least three of the gene products needed to assembly fibers.
I realized that I might be able to physically isolate the fiber components using ultracentrifugation. Jean Weigle, a senior research fellow who had accompanied Max Delbruck to Caltech, had an ultracentrifuge down the hall. Jean Weigle showed me how to generate sucrose density gradients, how to operate the centrifuge, and then collect fractionations afterwards. I then used the specific antibody to absorb the fractions. I believe my days of building model airplanes prepared me for the skilled manual operations needed for all the steps in the sucrose gradient centrifugation analysis. To this day I am surpised that I mastered that technology. I took no chances and collected hundreds of gradient fractions, each one assayed by a serum blocking assay, requiring hundreds of dilution tubes, hundreds of petri dishes. Years later I came to realize the value of the microbiological kitchen at Caltech which made all that possible.
I remember absolutely vividly the first experiment in which I identified a peak of phage proteins reacting with the anti-fiber antibodies. I posted the graph on the blackboard in the lab, and sent home a copy to my family, announcing that this result indicated I was going to succeed in my graduate work.
Using the specific antibodies combined with sucrose gradient centrifugation I was able to determine the multiple steps in T4 tail fiber assembly (King and Wood, 1969) – each half fiber was composed of a different protein, and three other proteins acted on the end of one, activating it for joining with the other.
Subsequently biochemist Bill Wood joined the Caltech faculty and Edgar and Wood demonstrated the striking result that infectious T4 could be assembled in vitro from lysates of mutants defective in assembly – in vitro complementation.
Electron Microscopy of Intermediates in Tail Assembly
From my exposure to Ernest Pollard at Yale, short stint with Alan Hodges in the Caltech EM lab, and learning of Kellenberger’s seminal studies, I had an appreciation of the value of Electron Microscopy. Michael Moody was a visiting postdoctoral fellow at Caltech from London, and generously instructed me in the use of the electron microscope, negative staining, and preparing carbon coated grids. I loved working in the cool, dark, air-conditioned EM room, and looking for phage substructures in lysates of mutant infected cells.
Among the most dramatic results were finding lysates of mutant infected cells which accumulated in addition to complete phage heads, what were clearly intermediates in phage tail assembly – free baseplates, baseplates with tail tubes, tails with tubes and sheaths, but not connected to the heads. The most interesting result was not explicit but was crystal clear – in the absence of baseplates, no tubes or sheaths were assembled. Clearly the pathway was strongly sequential. Since all the protein were synthesized at the same time, the sequential assembly had to represent conformational changed in the subunits driven by the assembly process itself. These exciting observations led me to analyze mutants in all the known T4 late genes, looking for such species. I returned to tail assembly at the MRC, and then at MIT.
NASA Antarctic Intermission with Charlie David and Roy Cameron
The late 1960s was the heyday of NASA and the preparation for the Moon landing. Already NASA was exploring a mission to Mars, in which the search for life on Mars would be a leading theme. A great deal of the program was housed in Caltech’s Jet Propulsion Lab. Toward the end of my thesis work I saw an announcement requesting grad students with microbiology training to go the Antarctic and characterize the microbial flora of the Dry Valleys off the Ross Sea. This was a cold desert, thought to resemble the surface of Mars. My Biology grad sidekick Charlie David also applied, and we convinced the PI soils scientist Roy Cameron to take us both.
We flew across the Pacific to New Zealand, and then from there on a ski-equipped C3 to the U.S. Antarctic Base at Ross Island in the Ross Sea. This was operated by the Navy and was a bevy of Quonset huts, including a laboratory.
We were taken out to Dry Valley sites by helicopter, and left there for a week or so. During that period, we took soil samples, environmental and weather measurements, and then returned to the base. The soil samples were plated on a variety of agar media, and we counted and examined the colonies formed. In general, they were not the expected psychrophiles, but bacterial generalists that were probably deposited by the winds. We also showed that though many samples were sterile, in very particular local environments, under transparent rocks, protected from the prevailing wind, soil algae probably provided nutrition for occasional bursts of bacterial growth. The publication of these experiments were my first scientific papers.
These results were used in the design of the microbial sampler for the Mars Viking lander, led by Caltech’s Professor Norman Horowitz.
We were the first humans to visit a number of the Dry Valleys, and so got to name them. As a result, the gazetteer of Antarctic names includes King Valley, David Valley and Lake Cameron. I’m still alternately proud and embarrassed in that matter.
Learning Labelling with Radioactive Amino Acids with Bill McClain and Sewall Champe at Purdue
I had been awarded a Jane Coffin Childs Fellowship to be a Postdoctoral Fellow at the MRC lab for Molecular Biology. However, no one had room for me in 1978. My stint working for the Jet Propulsion Lab ended, so I needed a position. Aaron Klug offered me a spot for 1979. Seymour Benzer and Sewall Champe had done the historic T4 rII gene analysis at Purdue, so I contacted them. Sewall Champe invited me to join his group. I didn’t know it at the time, but Benzer had already accepted a position in my department at Caltech, and had left Purdue.
I drove to West Lafayette Indiana, found an apartment on Main Street and set to work. Bill McClain and Nat Sternberg were the resident senior grad students and taught me how to label infected cells with radioactive amino acids, and then to determine incorporation into proteins.
My time at Purdue was limited by the absence of a microbiology kitchen that prepared sterile media and glassware, as was standard at Caltech. In addition, Sewall’s marriage had broken up, and he wasn’t present very much in the lab.
West Lafayette Indiana was in the midst of corn country and exemplified main line midwestern farming culture. Though the rest of the country was burning up in 1968, Purdue was voted the Greek (fraternity and sorority) Campus of the Year. But even at Purdue there was a small band of anti-war students, associated with the newspaper, and a few small but intense demonstrations were held. We had the sympathy of the Black football players, and I met Purdue star running back Leroy Keyes who went on to be an outstanding NFL player.
Electron Microscopy, Image Processing and more Phage Assembly at the Lab for Molecular Biology
In the spring of 1969 I moved to Cambridge, UK and took up residence on one end of the 3rd floor of the historic Hills Road Lab where Sanger, Watson, Crick, Brenner, Kendrew, Perutz, Huxley and Klug did their work. In that lab were Uli Laemmli who had come from PhD work on T4 head assembly with Kellenberger, as well as Joe Butler working on Tobacco Mosaic Virus assembly, and later Sidney Altman (subsequent Nobel laureate for RNA processing) and Jake Maizel, authority on structural proteins of DNA animal viruses, on sabbatical from Albert Einstein.
During this period Laemmli developed high resolution SDS gel electrophoresis. This was a major leap forward for molecular biology, allowing quantitative analysis of structural proteins, and a key method in the 50 years following. However, Aaron Klug in his autobiographical summary, leaves out any mention of this contribution, which was unrelated to his EM and Image Processing Nobel prize. I have written up a separate history of the development of SDS gel electrophoresis, which came from the intellectual collaboration of Laemmli and Maizel, assisted by my manual labor. My efforts to publish this have not been successful, so it follows as a separate appendix in this volume.
At the MRC Lab, Aaron Klug assigned David DeRosier to be my immediate supervisor. David gave me a book on Bessel functions and told me to come back and talk with him, once I had that under my belt. After a few days of struggling with Bessel functions, I decided that was not my future, and instead would take advantage of fact that LMB had a fully functioning microbiological kitchen for preparing media, Petri dishes, sterile pipettes, etc., left over from the T4 experiments of Brenner, Crick, and coworkers. So, I turned to focus on T4 tail assembly. Armed with Laemmli’s SDS gels we were able to identify the proteins encoded by all the genes required for T4 tail and tail fiber assembly. Laemmli had been very chary in his description of the high-resolution gels, stuffing the details into a figure legend. In King and Laemmli (1971) we described more clearly and more completely how to carry out the analysis. This paper remains my most cited publication.
Through working with Laemmli and Maizel on developing high resolution SDS gel electrophoresis, I had a much deeper understanding of how such gel electrophoretic separations worked, with and without detergent, than most protein biochemists. In later years at MIT I was able to put that to good use in sorting out features of tailspike folding, assembly and aggregation.
During this period Tony Crowther, Aaron Klug and John Finch were developing the image processing tools which would later transform electron microscopy. These required highly uniform and complex structures, to reveal their power. I provided the purified baseplates for the original application of rotational image processing, later extended to the expanded star configuration (Crowther et al, 1977).
Coming to MIT
I had been hired to fill the slot of Professor Cecil Hall, a pioneer of electron microscopy. I inherited an EM lab that had the first models built by Siemens and by RCA. It was almost a museum. Luckily Elaine Lenk, who ran the microscopes at the MRC, moved to Cambridge, USA, and she joined me as lead technician in charge of the microscopes. Early on I wrote a successful NIH grant to buy new microscopes, and we replaced the museum scopes with modern Phillips Microscopes.
All of the work below was publicly funded through primarily NIH grants and occasional NSF grants. Unfortunately, I don’t have access to the records, either of submissions, or awards, so don’t connect any of the accounts below to the grants which financed them. But I was always sharply aware that it was U.S. taxpayers who paid for our research.
The Biology Department had two cultures – an older biochemical and structural tradition represented by Gene Brown, Jack Buchanan, Francis Schmidt and Cecil Hall, and a newer molecular biology thrust represented by Salvador Luria, Boris Magasanik, Maurice Fox, Harvey Lodish, Ethan Signer, David Botstein and David Baltimore. (In fact, I had been hired for the structural biology position because I bridged the gap, with my PhD work in phage genetics!)
For the following 25 years we operated the EM lab as a service facility for any federally funded lab at MIT, as well as other groups in the Boston area. Skilled microscopist Erica Hartwig and Patricia Reilly followed in Elaine Lenk’s footsteps.
Identifying Each Genetically Determined Step in T4 Baseplate Assembly with Yoshiko Kikuchi, Sam Kayman, Peter Berget and Nadia Mykolajewycz
I had carried the complete collection of amber and temperature sensitive mutant strains from Caltech to the MRC and then back to MIT. Using Laemmli’s high resolution SDS gels, Nadia Mykolajewycz and I were able to identify all the polypeptide chains encoded by the “tail” genes (their mutant phenotypes were accumulation of complete heads).
Fuminori Yoshida showed that very minor but critical proteins needed to terminate the sheath and activate the tails for head binding, could be detected in a sucrose gradient using in vitro complementation. Unfortunately, Fuminori’s family needed him back in Japan, where he subsequently made significant contributions in the biotechnology industry.
Peter Berget joined the lab as a postdoctoral fellow and initially tackled the isolation of the two largest proteins of the Baseplate, the products of genes 10 and 7. We decided to move forward and sort out the steps in the assembly of the very complex baseplate. This was done by the very hard working and extremely careful experimentalist Yoshiko Kikuchi, who joined the lab as a postdoctoral fellow.
Yoshiko determined the sedimentation behavior of all the unassembled or partially assembled baseplate proteins accumulating cells missing one of the proteins. This allowed us to determine the baseplate assembly pathway, which had two components; assembly of the central hub, and assembly of the outer arms followed by their polymerization around the central hub (Kikuchi and King, 1975 a, b & c). The full tail assembly pathway is shown in Figure 1.
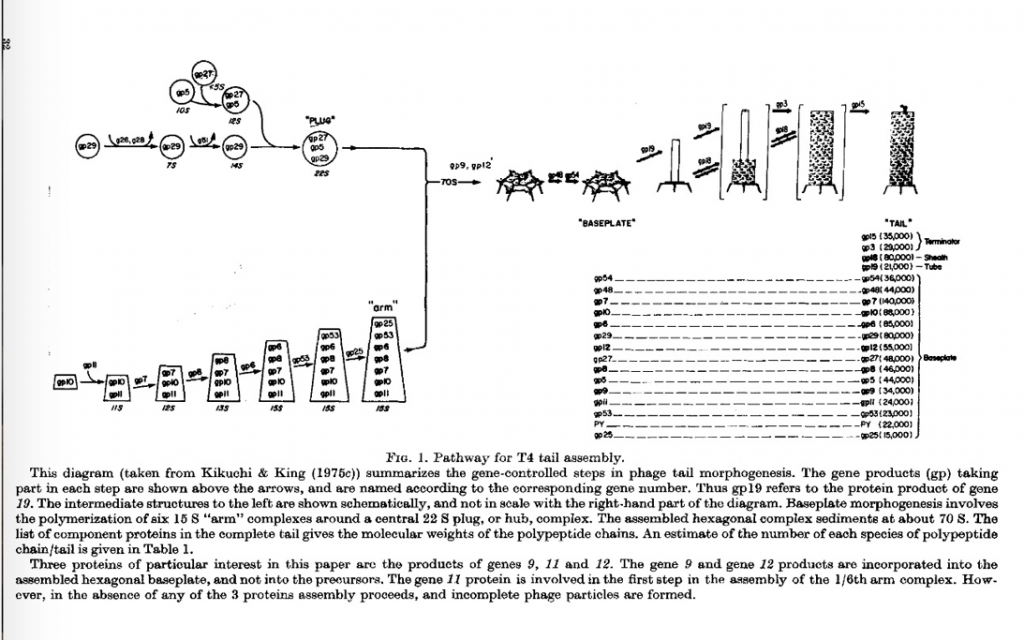
Yoshiko was also able to establish conditions in which these steps proceeded in vitro, by refinements of Edgar and Woods’ in vitro complementation between extracts of mutants infected cell. This confirmed the model above.
It should be noted that every one of her experiments demonstrated that the proteins required for the later steps in tail assembly, for example the tube and sheath subunits, remained in solution. As I noted above, to this day it is not recognized that all these proteins initially folded into a conformation in which they do not self-assembly. That is as sequential activation process, where subunits are bound on a growing structure, and conformationally activated to bind the next protein in the pathway.
During this period graduate student Sam Kayman set out to find direct genetic evidence of the interactions of the baseplate and tail proteins. He isolated and mapped second site suppressor mutations which identified many gene product interactions that hadn’t been identified previously. One of my failures as a mentor was not insuring that his papers from his thesis were actually submitted for publication. Sam was a perfectionist and always wanted to do one more experiment. I shouldn’t have gone along with that path. Sam died young, and we lost the opportunity for late publication.
David Botstein and P22
One of my fellow assistant professors was the very energetic microbial geneticist David Botstein, who carried out the T4 like conditional lethal analysis – isolation of amber and ts mutations – with phage P22 of Salmonella. David generated Salmonella strains carrying tRNA suppressors, as had been done with E. coli. P22 had distinctive advantages as an experimental system. There were no proteolytic cleavages involved in virus assembly, infected cells produced a high yield of particles, and the virions were very stable and maintained infectivity over long periods.
We decided to collaborate on the genetic analysis of P22 assembly and with the able assistance of Carol Waddell and Elaine Lenk produced two definitive papers (Botstein, Waddell and King, 1973; King, Lenk and Botstein, 1973) These suggested, but did not prove, that an empty shell was first produced and then filled with DNA.
Discovering Scaffolding Protein Function with Sherwood Casjens
At that time, I was joined by Sherwood Casjens. Sherwood had done his PhD theses at Stanford with pre-eminent phage geneticist Dale Kaiser, on phage lambda genetics and assembly. He was intellectually and technically prepared to take advantage of the opportunities afforded by the p22 system. Sherwood embarked on careful quantitative analysis using acrylamide gel electrophoresis of the involvement of the gene 8 scaffolding protein in P22 capsid assembly. (From familiarity with construction sites, and the transient role of scaffolding, I named the protein “scaffolding protein” which was helpful in making clear that it was absent from the mature virions.)
Sherwood’s experiment showed the quite extraordinary result that the scaffolding proteins had to be recycling during shell assembly, with each molecule participating in maybe half dozen rounds of shell assembly. Our paper was published in Nature (King and Casjens, 1974), though animal virologists remained loathe to accept such pathways as general. As this is written (2021) many animal virologist still fail to appreciate these steps in precursor shell assembly on the way to formation of mature virions. This is even more surprising given that the most effective HIV drugs act by inhibiting the protease whose cleavage activates early GAG polyproteins critical for steps in shell assembly.
Sherwood and I understood that the role of phage studies was a model for animal viruses that actually caused disease in the human population. In order to try to bridge the gap, we immersed ourselves in the animal virus literature and wrote a major review published in Annual Reviews of Biochemistry (Casjens and King, 1975). We predicted that double stranded DNA viruses such a as Adenoviruses and Herpesviruses, also assembled a procapsid with scaffolding-like protein. Very slowly over the subsequent 45 years, this result has been borne out.
Investigating Procapsid Assembly In Vitro with Minx Fuller, Peter Privilege, William Earnshaw, and Carol Teschke
Procapsids could be readily purified from cells infected with mutants blocked in DNA packaging giving us access to a key precursor in capsid assembly. When Minx Fuller joined the lab from Brandeis, she tackled the problem of obtaining coat and scaffolding subunits in a form in which they could be reassembled into closed shells in vitro. Minx was able to demonstrate the extraordinary result that isolated coat protein subunits isolated from the procapsid remained soluble and did not polymerize in the absence of scaffolding protein. Similarly, the isolated scaffolding subunits remained soluble in the absence of the procapsid conformation of the coat protein. That both were in biologically active conformation was clearly shown by their copolymerization into shells when mixed together (Fuller and King, 1980).
This extraordinary result showed that such structural subunits are not synthesized in the conformation in which they are found tightly bound together in the mature capsid, but in a precursor conformation which does not polymerize unless the right partners are available. This result is still not generally understood or appreciated in the biochemical, biophysical of virologic communities.
Peter Prevelige joined the lab at this time, with excellent training in physical biochemistry. He was able to characterize the kinetics of coat and scaffolding assembly in vitro, showing that the reaction was almost certainly a nucleation/polymerization reaction, in which subunits added predominantly to the growing shell (Prevelige, Thomas and King, 1993). This was the same result we had found with T4 tail assembly, with subunits synthesized in a non-polymerizing conformation, and polymerization and growth occurring on a growing structure, rather than in solution. Again, to this day the underling conformational switching nature of these reactions remains unrecognized and unappreciated.
Procapsid Structure with Bill Earnshaw, Barrie Greene and Wah Chiu
Once we knew that the scaffolding exited from the procapsid in association with DNA packaging, the question of its 3-D structure emerged. Ruth Griffin Shea had examined procapsids interacting with antibody against scaffolding, which looked like the scaffolding subunits exited throughout the shell. But those results didn’t have the resolution needed to influence structural biologists.
The first breakthrough came when Bill Earnshaw took purified procapsids to Harvard and collaborated with Steve Harrison, who had developed low angle X-ray scattering techniques for studying virus structure. These results indicated that the scaffolding protein was inside the shell of coat protein, and that the overall diameter of the procapsid was smaller than the mature virion, indicating expansion of the capsid lattice with exit of the scaffolding. This suggested a more dynamic procapsid to capsid transition than X-ray crystallographers expected.
One of the frustrations of working on T4 was the absence of high-resolution structures. Given the apparent true icosahedral symmetry of the procapsid, it seemed possible that it might crystallize. The leading lab in the country on X-ray structure of viruses was Michael Rossman, with his plant virus associate jack Johnson, at Purdue. Barrie Greene travelled to West Lafayette and spent enough time in Jack’s lab to generate Crystals of procapsids! I was thrilled. Sadly, over the Christmas holiday that year they were able to get synchrotron diffraction time, and found that the crystals didn’t diffract. To this day no one has obtained diffracting crystals of P22 or any other procapsid.
DNA Injection Proteins with Ruth Griffin Shea, Gerry Bryant and Edward Loechler
One of the most distinctive phenotypes of P22 mutants were those in genes 16 and 20. They accumulated morphologically normal DNA-filled particles, which were not infectious. Ruth Griffin Shea showed that these particles attached to cells in an apparently normal fashion.
Subsequently Jerry Bryant studied the strains in more detail, and found that the mutant particles could complement each other to produce a productive infection. This strongly suggested that they were injected into the infective cells (Bryant and King, 1984).
An aspect of these experiments utilized photo-inactivation of P22 particles with 9-amino acridine. This was model for the mechanism of action of photo-toxins. Ed Loechler joined the Lab as a postdoctoral fellow with a sophisticated knowledge of reaction kinetics. He was able to show that though 9-AA bound to DNA its action was to modify proteins bound to the DNA. It seemed likely that these included the products of genes 16 and 20.
The Recombinant DNA Controversy and the Cambridge City Council
During this period the Cambridge City Council was considering the possibility of regulating the newly developed gene cloning technologies. This was front-page news, and we were periodically visited by TV news teams, which was disruptive for people working in the lab. Ed Loechler and grad student Phil Youdarian were part of the group of Boston area microbial geneticists who believed the new technology was powerful enough to deserve regulation, so their presence was very supportive.
My own concern came out of my experience with occupational health and safety issues, and knowledge of the environmental and occupational basis of many human diseases. In addition, as someone on the inside, I knew that the opponents of regulation had already incorporated new startups, and were more interested in financial gain than in “unfettered” research. The few articles I was able to publish have hardly ever been referenced: King J. (1978) New diseases in new niches. Nature, 276(5683): 4-7. PDF at http://www.nature.com/nature/journal/v276/n5683/pdf/276004a0.pdf) and (King, 1978 – Journal of Infectious Disease). Despite the dozens of articles, books and videos on this controversy, the full story remains to be told, particularly the invisible role of financial investors in opposing regulation of the emerging technology.
This is symptomatic of the intense efforts of proponent of unregulated research to argue that there were no hazards involved. Luckily the general public recognized the need for increased lab safety and oversight, and eventually these were adopted nationally and internationally.
Elucidating Early Steps in Procapsid and Portal Assembly with Chris Bazinet, Carl Gordon, Maria Galisteo, and Barrie Greene
Early on we knew that the portal ring composed of gp1, did not form in the absence of scaffolding protein. A variety of creative studies of coat, scaffolding and portal mutants were carried out by Chris Bazinet, Carl Gordon and Barrie Greene.
Among the most surprising results were those of Chris Bazinet who found evidence supporting a model in which the scaffolding mRNA was a nucleator. We know from Sherwood’s studies that scaffolding protein interacted with its messenger. Chris was able to explain the bizarre result that some tailspike mutants could suppress procapsid assembly defects.
Carl Gordon and Barrie Greene embarked on extensive characterization of ts and cs mutants of the Coat (Carl) and Scaffolding (See Barrie’s chapter for a fuller account). These studies all supported intimate interactions of these proteins in both initiating and propagating procapsid assembly, as well as transformation to the native capsid after DNA packaging.
Political Interlude with Mel King and Jackie King
Boston’s 1981 Mayoralty election was the first one in which one of the candidates – Mel King – was African-American. Mel had been a State Representative, but also an adjunct faculty member at MIT. My wife Jackie was one of his Press Secretaries, an exciting and high-pressure position. I couldn’t help but also be involved. Mel lost, but the campaign generated a new voting constituency in Boston, that is continuing to influence local elections.
Soon after Jesse Jackson decided to run for the Democratic Nomination for President. Jackie was his New England press secretary and I was on the national campaign committee, the only scientist. Since then both of us have regularly served on Campaign Committees for School Committee candidates, City Council candidates, State Reps and Senators, and U.S. Reps and Senators. Often, I made a useful contribution in explaining the large but somewhat obscured role of federal NIH and NSF funding in supporting graduate students, faculty, and higher education institutions in general.
Revealing Atomic Level Capsid Structure with Wah Chiu and Team
Though the failure to obtain diffracting crystals of P22 procapsid or virions was discouraging, the gap was filled by the initiation of a long running collaboration with Wah Chiu. Wah was convinced that CryoEM was going to supplant X-ray diffraction for biological samples. He was the most focused, most selfless, and steadiest of all the scientists I had the honor of collaborating with.
Using highly purified procapsids prepared by Peter Privilege and Cammie Haase Pettengill, Wah was able to show even at low resolution that the procapsid shell had openings, which were closed in the mature virion. The movements of the capsid subunits in this transition were eventually revealed at atomic level.
The power of CryoEM was fully revealed when Peter Weigele prepared purified Phi 29 virions, in which the coiling of the tightly packed DNA was directly observed. That image was on the cover of Nature.
Wah took the writing of the papers as seriously as the work itself. I remember fondly travelling to Houston periodically where our co-authors team went collectively through draft manuscripts line by line.
When Peter Weigele and I decided to make the transition to studying phages of marine cyanobacteria, following the lead of MIT’s Penny Chisholm and Woods Hole’s John Waterbury, Wah immediately recognized the importance of these studies. He opened up a collaboration with Penny, on phages of Prochlorococcus and extended his team’s work to include our studies of marine phage infecting marine Synechococcus strains, as I describe in more detail below.
One of the most distinctive results was that even marine cyanophage assembled a precursor shell empty of DNA and then pumped the DNA into it. This suggested that all dsDNA viruses shared a common evolutionary origin.
In another section below, I describe our very productive collaboration in Wah’s Nanomedicine team, on the structure and function of the Human Chaperonin TRiC, or CCT, which is required for folding many essential mammalian proteins.
Temperature Sensitive Folding Mutants with David Goldenberg, Donna Smith, Ben Fane and Myeong Hee Yu
None of the hundreds of temperature sensitive mutants characterized by Bob Edgar and colleagues rendered the mutant phage particle sensitive to temperature. It was clear to us in the Edgar lab that the mutants must be acting at the level of phage assembly or earlier, chain folding. I had always been interested in large proteins like collagen and myosin and I didn’t believe for a second that the two stage transitions U<>N put forward for small proteins could account for the conformations of collagens, myosin or other complex proteins. Interfering with folding seemed much more likely. However, our Caltech group was unable to show that experimentally.
Peter Berget, who was characterizing the very large structural protein s of the T4 baseplate, became interested in P22. He found that the tailspike was expressed at high levels in P22-infected Salmonella and formed a very uniform and distinctive peak in sucrose gradients of P22 lysates. That suggested that the tailspike might provide a protein whose folding and assembly might be probed with P22 temperature sensitive mutants, which Botstein had isolated.
David Goldenberg tackled this project for his thesis. He showed unambiguously that tailspike proteins, once folded and assembled, were as thermostable as the wild type protein. This once again pointed to the intracellular folding and assembly pathways as the locus of the ts defect. Donna Smith, then our lab technician, carried out the other half of the study, showing that at high temperature the mutant chains were synthesized at wild type rates, but were not able to reach the native state. Unfortunately, the trimeric tailspike was a sufficiently unusual protein compared to insulin, RNASE, and hemoglobin, that it was easy for the results to be dismissed as an oddity of the particular protein utilized in the experiments. Subsequently we referred to these mutants as “tsf” for temperature sensitive for folding (Sturtevant et al, 1989).
During this period the revolutionary technology of DNA sequencing emerged. Myeong-hee Yu immediately recognized the opportunity, and mastered the then very demanding techniques and giant gels needed to determine the nucleotide sequences of DNA isolated from the P22 phage carrying ts mutations in the tailspike gene (Yu and King, 1988).
Keeping a Working Lab Working with Cammie Haase-Pettingell and Cindy Woolley
At both Caltech and the MRC Lab for Molecular Biology, I had learned the importance of the highly skilled research technicians that provided a foundation for a functioning laboratory. At MIT for many decades we all benefitted from the skills and attention provided by Cammie Haase Pettingell in maintaining strains, refining and transmitting experimental protocols, managing reagents and suppliers, and relationships with maintenance personnel. On the office side Cindy Woolley maintained the financial records, the website, and the Publication list found in the Bibliography. When grant deadlines loomed their attention to detail was essential for the many successful submissions. Of course Cindy also broadened our cultural horizons with her periodic flute recitals and chamber concerts.
Robert Seckler and team solve the 3-D Structure of the Tailspike
A leap forward occurred when Robert Seckler, who had been a student of Rudolph Jaenicke at Regensberg, grew crystals and solved the structure of the tailspike (Seckler et al,1989). Its main feature was three chains with long runs of beta- helix conformation. However, the single chains then wrapped around ech other forming a local triple beta helix. This explained both its high stability, and the necessity that the single chain folded precursors had to be in a different conformation than in the native state.
The beta-helix can be thought of as a very primitive protein fold, but one which generates a very well defined lateral surface. Seckler’s further studies on the complex with Salmonella lipopolysaccharide revealed that indeed it was this lateral surface – formed of residues from each of the three chains – that formed the extended LPS binding site.
Solving the Mystery of Inclusion body Formation with Cammie Haase-Pettengill, Margaret Speed, Scott Betts and Anna Mitraki
With the widespread development of gene splicing and cloning technology, biotech and pharmaceutical companies were moving rapidly to produce human therapeutic proteins in E. coli, such as insulin, growth hormones and many others. To the consternation of the chemical engineers charged with actual production of the protein of interest, recovery of soluble active protein was rare. Far more common was the accumulation of the polypeptide chains expressed by the cloned genes, in an aggregated, insoluble, and inactive state, named an inclusion body. This was completely mysterious at the time since biochemical and chemical engineers had been trained to view proteins with the (limited) tools of equilibrium thermodynamics, and if the chains were correctly synthesized, their analysis led to the belief that they would be in the native state – Anfinsen’s “lowest energy state.”
We knew that a significant fraction of at least the structural proteins of bacteriophage, could end up in such an aggregated state, for example coat proteins in the absence of the GroE chaperonins. In fact, the tailspike chains or carrying temperature sensitive amino acid substitutions, and synthesized at the high restrictive temperature, ended up in such an aggregated state. However, this literature was outside the world of main stream protein biochemistry and biochemical engineering.
The tailspike turned out to be invaluable for revealing the character of the inclusion body formation. It was so physiologically stable that there was no possibility the inclusion body state could be derived from denaturation of the native state. Cammie Haase-Pettingel showed this definitively by following the kinetics of inclusion body formation and showing that the aggregated chains derived from intracellular folding intermediates, rather than breakdown of the native state.
Though this result didn’t get much attention from the protein folding or protein biochemistry communities, the results percolated through the Biotech industry. This led to simple physiological approaches to increasing yield of native states, by for example growing cells at the lowest temperature supporting translation of the cloned proteins. Scott Betts showed this cold rescue convincingly for tailspike folding and maturation.
Another approach in biotechnology was to purify the very stable inclusion body state, which was already a predominantly chains of interest. Once these particles were isolated, they could be denatured and attempts made to refold in vitro. Of course, this often led to aggregation in vitro.
These off pathways but physiological processes were elucidated by creative experiments of Margaret Speed an MIT Chemical Engineering major, working with Professor Danny Wang. A leap forward occurred in Margaret’s very careful characterization of the competition between tailspike refolding and aggregation, both in vivo and in vitro. Using native gel electrophoresis, she was able to distinguish the multimeric intermediates along the aggregation from the intermediates leading to the native state.
Biochemists thought of aggregation as a non-specific process. But bio/pharma production engineers knew that the inclusion body was often a rather pure state of the over-expressed protein. Using mixtures of coat protein and tailspike we were able to demonstrate directly that the inclusion body pathways were specific, even when multiple species were aggregating (Speed, Wang and King, 1996).
Suppressors of Protein Folding Defects with Anna Mitraki, Ben Fane, Bob Villafane, and Cammie Haase-Pettingel
Once we understood clearly that ts mutants of the tailspike chains interfered with chain folding, it was reasonable to search for suppressor mutations of these ts phenotypes.
Graduate student Ben Fane, and postdoctoral fellows Anna Mitraki and Bob Villafane contributed to the isolation of such mutants.
Anna Mitraki, as noted above, had a deep understanding of the physiology of chain aggregation, as well as of the culture in the field. Her paper made a convincing case that folding defects and inclusion body pathways could be suppressed by single amino acid substitutions in the tailspike chain.
Tailspike In Vitro Refolding with Robert Seckler and German Colleagues
Whereas U.S. protein folding labs had focused on small polypeptide chains that exhibited two state folding and unfolding, European groups including Rudolph Jaenicke in Germany and Michel Goldberg in Paris had tackled important proteins in microbial physiology, often complex. Robert Seckler, a graduate student with Jaenicke recognized the potential of the tailspike. As noted above, he was able to solve the #-D structure revealing clearly the beta-helix underlying fold, and also to refold the denatured chains in vitro. This moved the tailspike from an obscure phage structural protein closer to the mainstream.
The intertwined triple beta-helical structure of the tailspike was quite extraordinary. Among many consequences, it revealed that the chains prior to coming together could not possibly be in the native state – the early precursor needed to have been in a different conformation. In addition we realized that the beta-helical fold provided a model for the amyloid deposits found in Alzheimer’s brains.
We enjoyed numerous visits across the Atlantic, and were always educated by interactions with Robert, who was a truly penetrating and persistent protein biochemist with deep knowledge of all aspects of biochemistry and microbiology.
He recognized that the straight triple helix of the tailspike provided the reactive surface for specific recognition and binding of the LPS molecules that were the tailspike substrates.
Raman Spectroscopy of Tailspike structure with George Thomas and Steve Raso
In the initial absence of a high-resolution X-ray structure, we desperately needed structural information on the tailspike. We contacted George Thomas, then at UMass Dartmouth and sent him purified sample. The Raman spectroscopy confirmed the predominantly beta sheet nature of the tailspike. When George moved to University of Missouri at Kansas City the collaboration became long term. His group also characterized Raman spectra of phage particles.
Subsequently a variety of indications called attention to the tailspike cysteines, which were reduced in the native state, but gave hints of unexpected properties. Linus Pauling had predicted that the cysteine SH groups would be hydrogen bonded in proteins, but such bonds were not resolved by X-ray spectroscopy in any protein. By producing mature tailspikes lacking each of the cysteines, Steve Raso produced a unique set of native, biologically active tailspikes.
George Thomas, working with the cysteine mutant strains obtained Raman spectra and was able to demonstrate that different cysteines H-bonds formed with different strengths resolvable in the Raman spectra. This was the first direct experimental measurement of the strength of such cysteine H-bond.
The tailspike cysteines had an additional distinctive life during chain folding, in which they formed transient S-S bonds needed to maintain the chains on the productive pathway, described in a section below
George spent a sabbatical year at MIT, where he proved to be one of the most good natured of our team, and was comfortable lowering his acutely sophisticated spectroscopes techniques, to learn the low-tech techniques for growing infected cells and purifying particles and proteins.
After retiring, George and his wife Martha – a long-time public-school teacher – established a summer home in Falmouth and Jackie and I spent many a lively dinner with them.
Probing Tailspike Using Antibodies with Bertrand Friguet, Cammie Haase-Pettingel, and Patricia Clark
As a result of being able to spend a sabbatical semester at the Pasteur Institute, we were able to tap their expertise with monoclonal antibodies. Bertrand Friguet led our instruction both from afar, and in a productive stay in the Lab.
Patricia Clark, who had also worked with Lila Gierash at UMass Amherst, was able to demonstrate using specific antibodies, that the conformation of the tailspike while still bound on the ribosome, was distinctly different from the final triple stranded beta helix.
Computational Approaches to Protein Folding and Capsid Assembly with Bonnie Berger, Lenore Cowen, Phillip Bradley, Sorin Istrail and Russell Schwartz
The twenty years from 1970 -1990 were a period of extraordinary increase in the power of computing. Though we had many inquiries from computer scientists with respect to collaboration, the productive collaboration emerged with Bonnie Berger and Lenore Cowan, initially MIT computer science grad students, later faculty at MIT and Tufts, and with Sorin Istrail then at Sandia national laboratory, and Russell Schwartz who joined the lab as a computational postdoctoral fellow (risky for him at the time).
Bonnie and Lenore recognized that if viral shells grew by one at a time addition of subunits to a growing shell, there might be a “Local rules” solution to modelling the process (rather than one traditional global equilibrium approaches). They were able to write algorithms which implemented rules for local subunit-subunit interactions, which generated a closed shell. We were very excited by the results, but unfortunately, there were few other experimental systems for describing the kinetics of shell assembly, making it difficult to assess the generality of the approach.
During this time the problem of the off-pathway aggregation of polypeptide chains competing with folding, had emerged as a serious problem in biotechnology, as described above. Bonnie suggested I attend the Rutgers Symposium on Discrete Math, where I encountered Sorin Istrail from the Sandia Supercomputer staff. They were modelling nuclear explosions, but with the fall of the Soviet Union, were looking for non-Cold War inspired projects. When Sorin asked me if I knew of any really hard projects in Biology, I responded that realistic modelling of protein folding, encompassing off pathway outcomes, was such a problem.
Sorin brought the problem back to his colleagues at Sandia, and also tapped Russell Schwartz for the work. Their breakthrough was instead of modeling a single sequence folding up, they modelled a pair which could diffuse and collide in space. Sticking together blocked productive folding and thus modeled aggregation. Even with the nation’s most powerful supercomputer, they could only handle two 14-residue chains, with two classes of side chains, hydrophobic and polar. However, that modeling revealed clearly the many pathways resulting in aggregation rather than productive folding.
During this time Bonnie’s group including Phil Bradley developed a very efficient algorithm for predicting beta helix conformation from sequence, “BetaWrap” (Bradley et al, 2001). The algorithm identified a small but distinctive family of elongated beta-helical proteins, which turned out to be primarily proteins involved in viral and bacterial binding to host cells. This was followed by more efficient and sophisticated versions, and was used by many in the exploring phage and microbial toxins and proteins.
Russell went on to tackle a range of questions which could be addressed by close analysis of distributions of amino acids. He also attended phage assembly meetings and became one of the rare computational biologists who also emerged himself in the wet results.
One of his results I found most satisfying was the suppression of long sequences of hydrophobic acids in soluble proteins, which we intuited followed from selection against aggregation during folding.
Transient Disulfide Bonds in Tailspike Folding with Susan Sather and Anne Robinson
Given the complexity of the tailspike, and the known role of GroE chaperonin in the folding of T4 phage proteins, it seemed reasonable to search for GroE chaperonin interactions. Susan Sather began examining the role of energy inhibitors as an approach. To our surprise she discovered that IAA blocked tailspike folding directly, demonstrating labeling of tailspike chains in vivo and in vitro with 14C-labelled IAA. This opened up a new line of investigation of the properties of the tailspike folding intermediates.
Soon after Anne Robinson, with a background in Chemical Engineering joined the lab as a postdoctoral fellow. She followed up by searching directly for transient S-S bonded tailspike folding intermediates. Despite considerable skepticism and hostility from referees from the biochemical community, she established the presence of such intermediates using nondenaturing gel electrophoresis.
It wasn’t until twenty years later that structural studies of other beta-helical proteins revealed the need for an autochaperone that initiated or organized the triple chain structure. These were cleaved off in other proteins but remained as the tailspike C-terminus. This domain acted as an auto-chaperonin for tailspike folding and assembly, and the transient disulfides functioned in that step.
Folding of Lens Crystallins and Cataract with Melissa Kosinski Collins, Ishara Mills Henry, Shannon (Flaugh) Thol, Ligia Acosta-Samson, Jeijin Chen, Kate (Drahos) Moreau, Fan Ron Kong and Nathaniel Schafheimer
The lab experience with aggregated proteins, and our experience with the Beta helical tailspike, a model of amyloid aggregates in mammals, suggested that it was time to begin direct study of some human disease whose pathology was associated with misfolding and aggregation. Rudoph Jaenicke had characterized the solution biochemistry of the gamma crystallins, and Christine Slingsby at Birkbeck College London had solved a number of crystal structure of these double Greek key Beta-sheet proteins. Professor George Benedek in MIT’s Physics Department had been studying lens proteins for many years. His research team included Ajay and Jayanti Pande, who recognized the value of being able to access purified lens proteins through cloning and expression in bacteria. We were able to assist with that effort, as well as provide electron microscopical visualization of their lens samples.
We then learned that single amino acid substitutions in the gene for gammaD -crystallin resulted in cataracts. That launched me into taking up the serious study of crystallin folding and unfolding, as a way of responding to the high incidence of cataract among aging populations.
The crystallins are extraordinarily stable and soluble and do not aggregate from their native states. Melissa Kosinski-Collins, well trained in her undergraduate work with Lila Gierash at UMass Amherst, tackled the study of the in vitro refolding and showed that the protein refolded in vitro at physiological temperature in the absence of chaperonins, despite competition from aggregation (Kosinski-Collins and King, 2003). Though there are no covalent reactions, it still took the chains many minutes to reach the native state even at 37oC. It was clear to us that achieving the highly stable Greek key fold was not kinetically easy. Upon denaturation in 5 M guanidinium hydrochloride (GuHCl) and subsequent dilution with buffer to initiate refolding, a robust aggregation reaction competed with productive refolding of the protein. That provided an initial in vitro model for cataract formation.
Melissa discovered that the fluorescence of gD-crystallin was hypersensitive to conformation, so that the fluorescence signal was a reliable and very sensitive reporter of the conformation of the protein. The aggregates had Trp fluorescence emission spectra that were intermediate between the native and denatured states, indicating that they might be composed of partially folded polypeptide chains.
Subsequent very careful and elegant physical biochemistry by Melissa, Shannon Flaugh, Ishara Mills Henry and Ligia Acosta-Sampson identified a partially folded precursor in which the C-terminal domain (C-td) was native-like but the N-terminal domain (N-td) is not fully folded. Furthermore, the isolated, aggregation-prone C-td of human γD-crystallin recruited the non-aggregating isolated N-td into off-pathway aggregates.
The multimeric alpha-crystallin chaperonin is a major protein of the lens, presumably protecting against protein denaturation, aggregation and cataract formation in situ. Ligia constructed no-Tryp alpha crystallins, which she employed to show that alpha crystallin recognized, bound and suppressed aggregation of the gD-crystallin chains (Acosta-Sampson and King, 2010).
Melissa subsequently constructed a series of crystallin genes in which three of four tryptophan’s were replaced by phenylalanine’s. These strains allowed very detailed analysis of the fluorescence absorption and fluorescence energy transfer operating within and between the Greek keys. Though direct determination of the conformation in the aggregated state had been elusive, molecular dynamics calculations by Zhou and colleagues of the IBM supercomputing group suggested a domain swapped interaction between partially unfolded crystallin chains.
Jeijin Chen pursued the fluorescence transfer and quenching reaction in the crystallins, and in collaboration with theoretical chemist Patrick Callis at Montana State, revealed the molecular mechanism of the quenching of the tryptophans through very rapid transfer of the energy to the backbone. The tryptophan/backbone conformations required for this were conserved in all crystallins with solved atomic structures. We interpreted this conserved quenching as a mechanism of protection of the retina, which very sensitive to UV; the crystallins present at high concentrations absorb the largest part of incident UV, but the quenching of the fluorescence protects them from absorption-induced denaturation.
During this period, we interested NMR spectroscopist Angela Gronenhorn in the crystallin project, and her new facility at the University of Pittsburgh produced high resolution NMR data that confirmed that perturbation of the native state was unlikely to be the mechanism by which mutations induced cataract in mammals.
The discovery that Alzheimer’s aggregates were amyloids, raised the question of the aggregated conformation in cataract. Katerina Papanikolopoulou, visiting from Greece, worked with an array of our partners and did indeed show that an aggregation pathway for human γD- and γC-crystallins led to formation of amyloid fibrils upon incubation at pH 3. (Katerina Papanikolopoulou 1, Ishara Mills-Henry, Shannon L Thol, Yongting Wang, Abby A R Gross, Daniel A Kirschner, Sean M Decatur, Jonathan King, 2008.)
Although the bulk of the aggregated protein removed from cataractous lenses is not in the amyloid form, it remains possible that amyloid fibers participate in initiation or early stages of cataract.
As a means of going deeper into the actual etiology of cataracts, Kate Drahos Moreau initiated a systematic study of mutants in mice which resulted in cataracts with the mouse lenses. Kate expressed lens proteins carrying the mouse amino acid substitutions, which were of buried residues. She discovered that they probably caused the cataracts by a mechanism we hadn’t considered, modification of the conformation that was not recognized by the alpha crystallin chaperonin.
We had tackled the crystallin/cataract project with the long-term goal of searching for small molecules that might inhibit aggregation, and provide a non-surgical cataract therapy. This was particularly attractive due to the prospect of delivering the putative drug as eye drops. Kate Moreau took this project on, and working with one of the Harvard Medical School Screening Facilities, set up a crystallin aggregation assay to search for compounds inhibiting the aggregation. Her impressive effort did indeed turn up “hits” deserving of further investigation. However, during that period we learned from Liliana Quintanar’s work (below) of the role of copper and zinc ions as inducers of crystallin aggregation. Many of the hits looked like metal binding compounds which gave us pause. In addition, Jason Gestwicki’s group at UCSF had discovered excellent candidates for inhibitors of crystallin aggregation including lanosterol. In hindsight, we probably shouldn’t have abandoned the search, but drug chemistry was far afield for our group.
Though we were focused on the UV absorption by crystallin tryptophans as protective for cataract, it was well known that this could be overwhelmed and UV irradiation in experimental animals and humans (through accidents) was well documented. Nathaniel Schafheimer took for his thesis project elucidating the mechanism of UV-induced aggregation. In fact, he showed that the tryptophan excited states transferred to one of the cysteines, which was activated to a free radical state, and subsequently irreversibly polymerized.
Subsequently Eugene Serebryany tackled directly the biochemical mechanism leading to off pathway aggregation of gD-crystallin. In an ingenious and penetrating series of experiments Eugene showed that a distinctive non-native disulfide bond intermediate, catalyzes domain swapping interactions leading to the domain swapped aggregated state (Serebryany and King, 2015). Computational modelling by his colleagues in the Shakanovich group support the mechanism.
All these results suggested that in the aging eye, depletion of chaperonin, and accumulation of damaged crystallins leads to aggregation by a domain swapping mechanism. depicting an overall model of cataract formation based on a population of proteins with partially unfolded conformations that preferentially aggregate, while this aggregation is suppressed in younger people, whose lenses still maintain adequate concentrations of the alpha-crystallin chaperonins.
Key Role of the Hydrophobic Buried Core and Sequential Beta-helix Folding with Ryan Simkovsky
The almost final breakthrough in understanding the tailspike folding pathway came from a heroic experimental analysis by Ryan Simkovsky. Ryan recognized a feature of the structure that we had all missed, which was that the hydrophobic residues formed long stacks that ran the length of the beta helical trimer. Scott Betts has shown earlier that some mutants of hydrophobic residues caused strong folding defects. Ryan mutated every one of the 100+ buried core residues in the tailspike sequence to alanine, and carefully characterized the resulting chains. To do this he generated a number of ingenious semi-automated experimental gel procedures, which enabled him to collect complete data sets on the more than 100 strains. (Unfortunately, these advances in equipment and methodology didn’t get properly documented, and are lost.)
Ryan’s results showed that disruption of any one of the great majority of buried core residues, prevented chain folding. The results pointed clearly to a model in which the sequential packing of the hydrophobics were critical for forming the turns of the Beta helix. Each turn essentially nucleated next turn. This explained why the tailspike folding and assembly was slow, many minutes per molecule. The model was consistent with all prior results, and deeply satisfying in physical and kinetic terms. Unfortunately, the single publication reported the results was probably insufficient to transmit the finding to the general protein folding community (Simkovsky and King, 2006 https://pubmed.ncbi.nlm.nih.gov/16505375/).
Zinc and Copper Binding in Cataract Formation with Lilliana Quintanar and Her Students
Upon receiving an invitation to speak at a Mexico/U.S. Protein folding symposium, I traveled to Mexico City, and got to know some of the excellent physical biochemists there. Among them was spectroscopist Lillian Quintanar on the staff of CINVESTAV (kind of NSF equivalent). Though it took a while our two groups eventually developed a very productive international collaboration. Eugene and Cammie visited Mexico, and Lilliana and her staff visited at MIT. Liliana was both an extremely gracious individual, but also a superior scientist – everyone loved having her in the Lab. Of course, she was burdened with the discrimination against women that operated on both sides of the Border, but she resisted marginalization and was a model for younger scientists.
Perhaps their most distinctive contribution was discovering the presence of specific binding sites on crystallin for copper and zinc, which led to distinctive aggregation pathways. These represented realistic models for occupational and environmental cataract among some populations (miners, smelters, etc.).
Characterizing Cyanophages of Marine Photosynthetic Synechococcus Cyanobacteria with Peter Weigele, Jacqueline Piret, Desislava Raytcheva and Claire Ting
Since fishing for flounder in Brooklyn’s Sheepshead Bay and on Long Beach Long Island, I had always been interested in marine biology. As the flounder disappeared from New England, including Cape Cod, I thought – I’m a biologist, I should be able to study some problem that was relevant for the marine environment. At some point I latched on to marsh grass, so important to Cape Cod ecosystems. But after some investigation I learned that no one had ever been able to grow marsh grass in the lab.
The Woods Hole Oceanographic Institute graduate program used MIT for its academic credentials. I volunteered for the Joint Committee, and for years spent a few afternoons a semester listening to WHOI grad students and faculty.
During about that time I saw a paper by marine microbiologist John Waterbury of Woods Hole faculty, which showed electron micrographs of phage which propagated on marine photosynthetic bacteria isolated from the open ocean. I was astonished that they looked just like T4, lambda and P22. Cleary there was my path.
I spent parts of the next two summers in John Waterbury’s lab, trying to absorb some of his experience and expertise.
At MIT, Penny Chisholm in Earth, Atmospheric and Planetary Science, had opened new ground with her discovery of the widespread marine photosynthetic Prochlorococcus, and was interested in phages infecting Prochlorococcus. We tried to join in that effort, but sadly, Penny viewed us as competitors rather than collaborators.
So, we shifted gears back to the Synechococcus strains and phages that John Waterbury had characterized. And Peter Weigele, Welkin Pope and Cammie were able to develop a protocol for growing these very particular open ocean micro-organisms to high titer in aerated high light cultures. Introduction of certain of the Waterbury phages lysed the cultures! This opened the way for a full characterization of cyanophages infecting Synechococcus strains. Peter Weigele described the infections by Syn9 and Welkin Pope by Syn5. Our long-time phage lambda colleague at the University of Pittsburgh, Roger Hendrix, arranged to have the genomes sequenced.
Professor Jacqueline Piret took a sabbatical in the lab, and working with Cammie was able to develop a plaque assay for Syn5, which opened up access to the intracellular kinetics of cyanophage assembly.
Jacqueline’s Northeastern grad student signed onto the project and was able to isolate procapsids from Syn5 infected cells and show that these phages – which must have evolved far earlier than phages of enteric bacteria, followed the same general pathway – assembly of a precursor shell empty of DNA, into which DNA is actively pumped, and then closed to form an infectious virion. Since this was known to be the pathway for adenoviruses and herpesviruses the result argued that the mechanism of capsid assembly and dsDNA packaging evolved once during evolution in the marine environment, and that all subsequent dsDNA phages and viruses utilize this mode.
Entering The World of Human Chaperonins with Kelly Knee , Oksana Sergeeva, Wah Chiu, Greg Pintilie and Dave Gossard
As Wah Chiu shepherded the increased resolution of CryoEM, he also worked to expand its applications. One of his most productive efforts was bringing together a very diverse and strong-minded group of collaborators to form the Center for Protein Folding Machinery, financed by an NIH Nanomedicine Center Grant. He recruited me, and I brought Postdoctoral Fellow Kelly Knee, and graduate student Oksana Sergeeva to the team. Computer 3-D design wizard Dave Gossard from Mechanical Engineering also joined with his student Greg Pintilie. Judith Frydman at Stanford was the chaperonin anchor, but we were able to make significant contributions. This was in part because the in vitro folding of the human crystallins described above provided a realistic substrate to characterize mammalian chaperonin function in vitro.
After initial work on the prokaryotic MMCPN complexes, we decided to tackle the actual human chaperonin complex, known as TRiC or CCT (Cytosolic Chaperone for Tubulin). This double ring structure had 8 protein subunits encoded by 8 different genes. Kelly Knee developed a labor-intensive protocol for purifying human CCT from Hela Cells, and with Oksana generated bacterial expression systems for each of the 8 CCT genes.
A leap forward occurred when Oksana discovered that CCT4 and CCT5 subunits formed oligomeric rings in E. coli. This led to high resolution studies with our colleagues Paul Adams, Pereira and Ralston at UC Berkeley.
Oksana went on to characterize mutant subunits associated with neuropathies in humans, an exciting connection between basic biomedical research and human disease. The various results certainly justified the Nanomedicine program investments.
Service to the Biomedical Research Community
From my first experience attending the Lake Arrowhead and Cold Spring Harbor Phage meetings, I realized how important these face-to face meetings were to the development of the fields. I spent considerable time supporting such conferences, or bringing them into existence. Through invisible efforts in obscure committees I helped give birth to the FASEB Summer Conferences on Virus Assembly, and the FASEB Summer Conference on Protein Folding in the Cell. Within the American Society of Microbiology, I initiated the Bacteriophage Section (M) of the Society.
Probably the most significant of such efforts was the collaboration with Lila Gierash to move protein folding from a narrow silo only accessible to a select group of physical biochemists, to its full significance as a fundamental biological process influencing all cells. We organized a Symposium at the interdisciplinary American Association for the Advancement of Science meetings – “Deciphering the Second Half of the Genetic Code”. This was a very different formulation that the somewhat sterile two state thermodynamic description of PP chain folding. In those days AAAS meetings were well tended by a wide range of scientists, rather than science journalists, the present attendees. Every seat was filled, and extra rooms had to be opened. Lila and I invited scientists who were investigating the full range of protein folding, misfolding and chaperonins.
This led to Chemical and Engineering News inviting a significant Review (King, 1989). In addition, Lila and I set about recruiting authors for a volume that could be used in teaching, also titled “Deciphering he Second Half of the Genetic Code” by Gierash and King). This was a truly superior volume, in which we had harangued the authors to write for a general scientific reader, rather than their in group. Unfortunately, soon after its publication, AAAS decided to go out of the publishing business, and it went out of print after early sales of 1000+ volumes. That ended my involvement in trying to publish books.
MIT Teaching and Curricula Development
I always found that teaching was essential to my own intellectual development, because of the need to try to understand as fully as possible what was known about a much wider range of experimental system than one dealt with in the lab.
I taught very successful course in Electron Microscopy for Juniors and Seniors, the Development Section of our Introductory Laboratory Course for Freshmen and Sophomores, and The Protein Folding Problem, and Protein Folding and Human Disease (the latter with Susan Lindquist) for advanced undergraduates and graduate students. I taught the latter classes for decades, with very positive feedback from students. Instead of a final exam, the students researched and wrote an u to date report on a key protein or problem, and presented their findings to the class. Students loved the opportunity to do a serious research paper, and then to present to their classmates.
Sadly, at no time did any member of my department ever attend any of the classes, inquire about my teaching innovations, or otherwise pay any attention. (Except in one semester in which Phil Sharp was Department Chairman, he did read my evaluations and comment positively.) All the Biology Department Chairs – Boris Magasanik, Gene Brown, Maurice Fox, Gene Brown, Bob Sauer, Chris Kaiser, Tania Baker and Alan Grossman believed in maintaining quality teaching, but none of them had the pedagogical training or experience needed to implement improvements as they were needed. However, the general value system did generate an environment in which most faculty did their best.
Public School Curricula Development with Melissa, Ishara and Lisa Guisbond
In the mid-1980s the Commonwealth of Mass Secretary of Education, John Silber, introduced high stakes MCAS tests into the public-school system. This was part of the generally right-wing effort to narrow and limit curriculum, dumb down public education, limit democratic control of curricula, and increase avenues for corporate profit from public education. Our sons Aaron and Andrew were then in elementary and middle school, and were among the hundreds of thousands of public-school students negatively impacted by the backward policy. Jackie and I became active with the thousands of parents and teachers across the Commonwealth who opposed the high stakes testing. We helped form the Massachusetts Coalition for Authentic Reform in Education, and Jackie became one of the statewide organizers, with Larry Ward and Lisa Guisbond, travelling around the state meeting with parents, teachers and students. As a result, I became quite knowledgeable and engaged with the state of public education in Massachusetts.
In 2008, as part of the economic stimulus, Obama directed some supplementary funds to the NIH. We wrote and were awarded a science teaching and curricula development grant. Melissa Kosinski Collins, Ishara Mills-Henry and Lisa Guisbond then set about developing a superior “Science of the Eye” curriculum modules for middle schools and high school science teachers. They offered summer workshops, visited classrooms, and brought exciting scientific advances in an area that engaged the students – vision. Sadly, when the grant expired, we were unable to continue the program. However, it has been gratifying that both Melissa and Ishara are leading teaching faculty at their institutions, and Lisa is Executive Director of Citizens for Public Schools, which carried forward the opposition to MCAS. Similarly Jackie, Aaron and Andrew continue to be active in the promotion of quality and equitable education, through Citizens for Public Schools and other community organizations.
From Biomedical Research to Nuclear Disarmament
At Yale I joined demonstrations opposing the development of submarine-launched nuclear missiles. From that time on I was active in efforts to end the War in Vietnam, oppose the development of biological weapons, and reduce the risk of nuclear war. This eventually took the form of Chairing the annual MIT “Reducing the Threat of Nuclear War” Conferences, and becoming heavily involved in Mass Peace Action, one of the oldest and largest anti-war groups in the nation. My own focus has always been on the enormous costs of these weapons, and the draining of human needs programs to finance them.